



版权说明:本文档由用户提供并上传,收益归属内容提供方,若内容存在侵权,请进行举报或认领
文档简介
1、第十三章自身免疫与器官移植从前面的章节我们已经了解到获得性免疫应答是宿主抵抗外来感染的重要防御机制,对维持机体的健康起着重要的作用。但不幸的是,有时获得型免疫应答清除的抗原不是感染原的,此时往往引起严重疾病。这种免疫应答与针对感染原的获得性免疫在机制方面基本相同,所不同的只是抗原。在第十二章,我们介绍了环境中的抗原是如何引起过敏性疾病和其他超敏反应的。这一章,我们将介绍针对这两类特别重要的抗原所产生的免疫应答:一类是针对自身组织抗原的自身免疫(autoimmunity),这种免疫应答会导致以组织损伤为特点的自身免疫病(autoimune diseases);第二类是针对移植器官的免疫应答,即移
2、植排斥(graft rejection)。我们将进一步探讨这些疾病的发展进程,以及导致这些不良获得性免疫应答的机制。自身免疫直接针对自身抗原 自身免疫病是针对自身抗原的获得性免疫反应。针对外来抗原的获得性免疫反应的正常效应是清除外来抗原。例如,杀伤性T细胞清除病毒感染的细胞,可溶性外来抗原形成抗原抗体复合物后被单核巨噬系统(如巨噬细胞)清除。但是,当自身抗原引发获得性免疫反应后,由于免疫效应系统不可能将这些自身抗原完全清除,就会产生持续的免疫反应,引起组织的慢性炎症损伤,这种损伤甚至有可能是致死性的。自身免疫引起组织损伤的机制与保护性免疫和高敏反应在本质上是相同的。表13.1列出了几种常见的自
3、身免疫病。我们知道获得性免疫反应是由抗原特异性T细胞激活的,而自身免疫反应的激活也是如此。由此,自身抗原特异性T细胞就会直接或间接地引起组织损伤:细胞毒性T细胞和TH1异常激活的巨噬细胞直接引起广泛的组织损伤;异常的T辅助细胞可使自身反应性T细胞产生自身抗体。自身免疫是T细胞和B细胞受体高度多样性的结果,这种高度多样性确保它们可识别几乎所有的病原体。虽然在成熟过程中,大多数与自身抗原高亲和力的受体被清除,但仍有一些低亲和力的自身抗原反应性受体被保留下来。虽然现在还不清楚自身免疫是怎么发生的,但是环境因素和遗传因素,尤其是MHC的基因型非常重要。一过性的自身免疫广泛存在,但是只有持续的自身免疫引
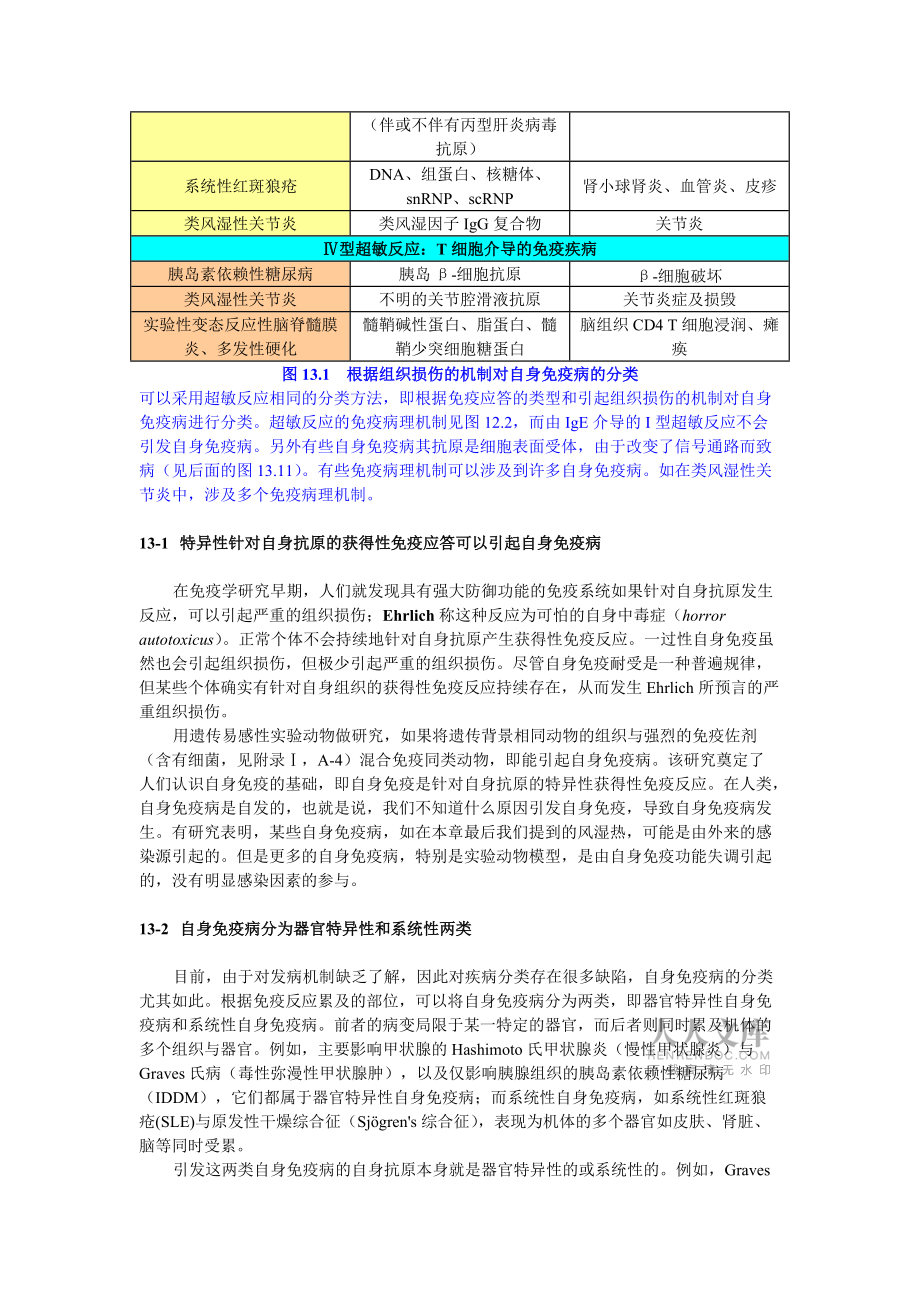
4、起组织损伤后才有临床意义。在这一部分,我们将探究自身免疫的类型以及它引起组织损伤的机制。在本章的最后一节,我们将探讨自身免疫耐受缺失和自身免疫反应启动的机制。根据免疫病理机制对自身免疫病进行分类症状自身抗原结果型超敏反应:针对膜表面或基质抗原的抗体自身免疫性溶血性贫血Rh血型抗原,I抗原由补体及FcR阳性的巨噬细胞导致红血细胞破坏,贫血Goopasture氏综合症血小板整合素,Gpb:a异常出血寻常性天疱疮上皮钙粘蛋白表皮内大疱急性风湿热链球菌细胞壁抗原,与心脏肌肉组织交叉反应的抗体关节炎,、心肌炎,心脏瓣膜瘢痕型超敏反应:免疫复合物介导的疾病原发性冷球蛋白血症类风湿因子,IgG复合物(伴或不
5、伴有丙型肝炎病毒抗原)系统性血管炎系统性红斑狼疮DNA、组蛋白、核糖体、snRNP、scRNP肾小球肾炎、血管炎、皮疹类风湿性关节炎类风湿因子IgG复合物关节炎型超敏反应:T细胞介导的免疫疾病胰岛素依赖性糖尿病胰岛-细胞抗原-细胞破坏类风湿性关节炎不明的关节腔滑液抗原关节炎症及损毁实验性变态反应性脑脊髓膜炎、多发性硬化髓鞘碱性蛋白、脂蛋白、髓鞘少突细胞糖蛋白脑组织CD4 T细胞浸润、瘫痪图13.1根据组织损伤的机制对自身免疫病的分类可以采用超敏反应相同的分类方法,即根据免疫应答的类型和引起组织损伤的机制对自身免疫病进行分类。超敏反应的免疫病理机制见图12.2,而由IgE介导的I型超敏反应不会引
6、发自身免疫病。另外有些自身免疫病其抗原是细胞表面受体,由于改变了信号通路而致病(见后面的图13.11)。有些免疫病理机制可以涉及到许多自身免疫病。如在类风湿性关节炎中,涉及多个免疫病理机制。13-1 特异性针对自身抗原的获得性免疫应答可以引起自身免疫病在免疫学研究早期,人们就发现具有强大防御功能的免疫系统如果针对自身抗原发生反应,可以引起严重的组织损伤;Ehrlich称这种反应为可怕的自身中毒症(horror autotoxicus)。正常个体不会持续地针对自身抗原产生获得性免疫反应。一过性自身免疫虽然也会引起组织损伤,但极少引起严重的组织损伤。尽管自身免疫耐受是一种普遍规律,但某些个体确实有
7、针对自身组织的获得性免疫反应持续存在,从而发生Ehrlich所预言的严重组织损伤。用遗传易感性实验动物做研究,如果将遗传背景相同动物的组织与强烈的免疫佐剂(含有细菌,见附录,A-4)混合免疫同类动物,即能引起自身免疫病。该研究奠定了人们认识自身免疫的基础,即自身免疫是针对自身抗原的特异性获得性免疫反应。在人类,自身免疫病是自发的,也就是说,我们不知道什么原因引发自身免疫,导致自身免疫病发生。有研究表明,某些自身免疫病,如在本章最后我们提到的风湿热,可能是由外来的感染源引起的。但是更多的自身免疫病,特别是实验动物模型,是由自身免疫功能失调引起的,没有明显感染因素的参与。13-2 自身免疫病分为器
8、官特异性和系统性两类目前,由于对发病机制缺乏了解,因此对疾病分类存在很多缺陷,自身免疫病的分类尤其如此。根据免疫反应累及的部位,可以将自身免疫病分为两类,即器官特异性自身免疫病和系统性自身免疫病。前者的病变局限于某一特定的器官,而后者则同时累及机体的多个组织与器官。例如,主要影响甲状腺的Hashimoto氏甲状腺炎(慢性甲状腺炎)与Graves氏病(毒性弥漫性甲状腺肿),以及仅影响胰腺组织的胰岛素依赖性糖尿病(IDDM),它们都属于器官特异性自身免疫病;而系统性自身免疫病,如系统性红斑狼疮(SLE)与原发性干燥综合征(Sjgrens综合征),表现为机体的多个器官如皮肤、肾脏、脑等同时受累。引发
9、这两类自身免疫病的自身抗原本身就是器官特异性的或系统性的。例如,Graves氏病是针对甲状腺组织的甲状腺刺激激素(TSH)受体产生抗体,Hashimoto氏甲状腺炎是针对甲状腺的过氧化酶,I性糖尿病是针对胰腺的胰岛素。系统性自身免疫病,如系统性红斑狼疮,在血循环中存在抗核抗体以及抗RNA剪切体的抗体,相应的抗原存在于几乎所有的有核细胞。器官特异性自身免疫病与系统性免疫性疾病的发病机制可能不同,这也许是自身免疫病分类的生物学基础。此外,这类疾病存在个体和家族聚集性,也从另外一个角度证明这种分类的正确性。器官特异性自身免疫病可以以不同形式发生于同一个体,如我们经常发现病人同时患有自身免疫性甲状腺病
10、与自身免疫性白癜风。同样,系统性红斑狼疮与原发性干燥综合征(Sjgren氏综合征)也可同时发生于一个患者或同一家族的不同成员。自身免疫病的聚集倾向有助于对它们进行进一步分类,每一亚类都有不同的发病机制。图13.2是自身免疫病常用的亚类分型表。通过该表可以看出器官性和系统性的分类不是绝对的,例如自身免疫性溶血性贫血即可局限于一个特定的器官,也可以与系统性红斑狼疮并发而作为系统性自身免疫病的一部分。从理论上说,每个人都有罹患自身免疫病的可能,但实际上自身免疫病只存在于某些易感人群。下面我们首先讨论自身免疫病的易感因素。器官特异性自身免疫病型糖尿病Goopastures综合症多发性硬化Graves病
11、Hashimotos甲状腺炎自身免疫性Addisons病白癜风重症肌无力系统性自身免疫病类风湿性关节炎硬皮病系统性红斑狼疮原发性Sjogrens综合症多发性肌炎图13.2根据“器官特异性”和“系统性”对常见的自身性免疫病分类有聚集倾向的自身免疫病被列在同一方框。所谓聚集倾向是指几种自身免疫病影响同一个人或同一家族的不同成员。不是所有的自身免疫病都可这样归类。例如,自身免疫性溶血性贫血即可独立发生又可与系统性红斑狼疮(SLE)并发。13-3 环境因素和遗传因素,特别是MHC基因影响自身免疫病的易感性通过对家族性自身免疫病,特别是双胞胎自身免疫病的研究,可以发现遗传易感基因。最常用的方法是半定量比
12、较单卵受精与双卵受精的双胞胎罹患自身免疫病的几率。也就是说,如果某一自身免疫病在两种双胞胎人群中发病机会都很高,那么该病可能与他们的周围环境和遗传背景都有关。但是,如果单卵受精双胞胎人群发病几率明显高于双卵受精者,那么遗传因素可能比环境因素更重要。目前,对多种自身免疫病(如IDDM、类风湿性关节炎、多发性硬化病和SLE等)的研究都在进行。在发病的双胞胎人群中,有20%的单卵双胞胎同时发病,而双卵双胞胎人群的发病率却不足5%。有研究者用同样的方法比较糖尿病患者同胞人群与一般人群的糖尿病的发病率,虽然家庭成员的相同生活环境也是发病的一个因素,但两者的差异也能反映遗传因素在疾病发生过程中的作用。对双
13、胞胎和家族性自身免疫病研究显示,环境因素与遗传因素在该类疾病的起始过程起着重要的作用。另外发现,有些品系的小鼠几乎都会发生自发的或实验诱导的自身免疫病,而其它品系的小鼠则不存在这种现象。这些发现促使人们去努力寻找自身免疫病的易感基因。目前,大家一致认为自身免疫病与MHC基因型有关。图13.3显示了自身免疫病与人白细胞抗原(HLA)基因型的关系。大多数自身免疫病与MHC II类分子的等位基因型有关。个别情况下,也与MHC I类分子的等位基因型有关。HLA血清型与自身免疫病易感性的关系疾病HLA等位基因相对危险性性别比(:)强直性脊柱炎B2787.40.3急性前房黑色素炎B27100.5Goodp
14、asture综合症DR215.91多发性硬化DR24.810Graves病DR33.745重症肌无力DR32.51系统性红斑狼疮DR35.81020IDDMDR3/DR4杂合子251类风湿性关节炎DR44.23寻常性天疱疮DR414.41Hashimotos甲状腺DR53.245图13.3HLA血清型、性别以及自身免疫病易感性之间的关系自身免疫病HLA等位基因的相对危险度是指自身免疫病HLA等位基因携带者数除以预期的该人群中HLA等位基因携带者数。该人群中预期的HLA等位基因携带者数目根据一般人群中HLA等位基因出现的频率计算,实际上是与HLA-DQ等位基因有关,后者与DR基因密切相关,这不能
15、用血清学方法检测到。有些自身免疫病有明显的性别差异,提示性激素参与该病的病理过程。与此相一致的是,性别差异的比例在初潮后与绝经前这个年龄段最大。最初,自身免疫病与MHC基因型的关系是通过比较患病人群与正常人群中不同等位基因的出现频率来确定。通过这种方法发现了IDDM与等位基因HLA-DR3和HLA-DR4有关(见图13.4)。使用同样的方法也证明了MHC II类分子的等位基因HLA-DR2对IDDM具有显性保护作用,即在含有HLA-DR2等位基因的个体,即使有其它易感基因的存在,也极少发生糖尿病。另一种确定MHC基因在自身免疫病中是否重要的方法是研究患者的家属,结果发现两个同胞罹患同一种自身免
16、疫病的几率远远高于预期,因为他们有着相同的MHC单倍型(haplotypes)(见图13.5)。健康对照图糖尿病图图13.4IDDM的易感性与HLA基因型的关系通过血清学分析表明,糖尿病人群的HLA基因型(下图)与正常人群(上图)不同。几乎所有的糖尿病患者都表达HLA-DR3和/或HLA-DR4,并且HLA-DR3/DR4杂合子出现的频率远高于正常人群。这些等位基因与决定IDDM易感性的HLA-DQ基因密切相关。在糖尿病人群几乎没有发现豁免IDDM的HLA-DR2基因。X代表除DR2、DR3、DR4以外的其它等位基因。关于胰岛素依赖性糖尿病HLA基因型的家系研究同胞的患病的同胞数没有相关性HL
17、A的预计数(图)(图)拥有2个相同的HLA等位基因拥有1个相同的HLA等位基因拥有0个相同的HLA等位基因拥有2个相同的HLA等位基因拥有1个相同的HLA等位基因拥有0个相同的HLA等位基因图13.5家系研究表明IDDM易感性与HLA基因型有关当两个或两个以上的同胞罹患IDDM,就可以比较患病同胞的HLA基因型。患病同胞具有两个相同的HLA单倍型远高于预期。对HLA基因型测序能够比血清学方法更准确地确定自身免疫病与HLA基因型的关系。例如,现在认为IDDM与DR3和DR4等位基因的相关性是由DQ基因引起的。DQ氨基酸序列中特定部位氨基酸的多态性与IDDM的发病显著相关。 在多数情况下,DQ第5
18、7位的氨基酸是天冬氨酸,它具有跨越DQ分子的肽结合缝隙而形成盐键的能力。在白种人糖尿病患者中,57位的天冬氨酸经常被替换为缬氨酸、丝氨酸或丙氨酸,导致DQ分子形成盐键的能力丧失(见图13.6)。非肥胖型糖尿病(nonobese diabetic,NOD)小鼠是一种自发发生糖尿病的小鼠,它与DQ同源的MHC II类分子的第57位是丝氨酸,称为I-Ag7。DQ链57位影响IDDM的易感性(图)链,57位,链与IDDM抵抗有关(图)与IDDM易感性有关(图)图13.6MHC类分子氨基酸序列改变与糖尿病易感性的关系大多数人的HLA-DQ1链第57位是天冬氨酸。而在白种IDDM病人中,该位置经常是缬氨酸
19、、丝氨酸、丙氨酸或其它氨基酸。如DQ分子骨架结构图所示,DQ1链第57位天冬氨酸与临近的链(灰色)的精氨(粉红色)酸形成盐键(中间图绿色)。当替换为其它不带电荷的氨基酸(如丙氨酸,最下面图黄色所示)时就破坏了这种盐键,从而改变了DQ分子的稳定性。在自发糖尿病的NOD小鼠,其同源分子1-A57位是丝氨酸。通过转基因技术,将链57位替换为天冬氨酸,能明显降低NOD小鼠糖尿病的发生率。该图由C.Thorpe授权。T细胞与特定抗原的反应依赖于MHC的基因型,且T细胞参与了自身免疫反应,因此我们不难理解MHC基因型与自身免疫病有关。人们推测,不同等位基因表达的MHC分子将自身抗原递呈给自身反应型T细胞的
20、能力可能不同。这种假说与T细胞参与自身免疫病的事实一致。例如,已发现MHC I类和II类分子参与了糖尿病的发生,与已知的CD8与CD4阳性T细胞都参与该病相吻合。我们知道MHC I类和II类分子分别向CD8与CD4阳性T细胞递呈抗原。另一个将MHC与自身免疫病易感性联系的假说强调MHC分子重塑T细胞受体的能力(见第七章)。这种假说认为与MHC分子结合的自身抗原肽能够促进发育中的胸腺T细胞的阳性选择,这些胸腺细胞对自身特定抗原产生反应。由于自身反应性抗原肽表达量很低或者与自身的MHC分子结合能力很弱,使自身反应性T细胞不能发生阴性选择。但这些T细胞与其它MHC分子结合后,可以发生富集,促进阳性选
21、择。在NOD小鼠中,由于MHC II类分子中存在I-Ag7,使得它与多种自身抗原肽的结合能力都很弱,因而削弱了自身反应性T细胞的阴性选择,这种现象也支持上述假说。但是,自身免疫病的遗传易感性不仅仅由MHC基因决定。研究发现,具有完全相同基因的同卵双生子比MHC基因相同的亲属罹患同一种自身免疫病的几率更大,说明MHC以外的遗传因素参与了自身免疫病的发生。最近,对人类和小鼠的自身免疫性糖尿病进行遗传学分析发现了MHC以外的独立遗传因素参与了该病的发生。胸腺内的自身抗原量也可以影响自身免疫病的发生。例如,IDDM患者的自身抗原是胰岛素,由于位于胰岛素基因上游微卫星DNA的多态性,导致不同个体胰岛素的
22、表达水平发生差异。胰岛素水平表达高的个体罹患IDDM的几率小,而表达水平低的个体患病率高,这是由于胸腺内高水平的胰岛素可以使针对胰岛素反应的T细胞消失。13-4系统性红斑狼疮易感基因为其病因学的研究提供重要线索SLE最显著的异常是血清中出现针对细胞自身抗原(如染色体)的抗体。这种针对自身抗原的免疫耐受是如何打破的呢?对人类和小鼠的研究显示,一系列基因参与了SLE的发病,根据它们的生理功能分为三类(见图13.7)。第一类基因的产物清除产生自身抗原的死细胞或垂死细胞。将这一类基因中的四个在小鼠中分别敲除可以造成SLE的动物模型(见附录I,A-47)。第一个基因是编码补体蛋白C1q的基因,它与其它的
23、补体蛋白共同作用参与免疫复合物和凋亡细胞的清除。第二个是血清淀粉样P物质基因,它编码的蛋白可以与暴露的染色质结合而不被免疫系统识别,敲除的结果是产生针对染色体的自身抗体,以及循环免疫复合物在肾脏沉积,从而引起肾小球肾炎。第三个基因是DNase I基因,它能消化细胞外染色体,敲除的结果与第二个基因相同。第四个基因是免疫球蛋白链基因,敲除的结果导致分泌型IgM缺乏,也会产生同样的表型。分泌型IgM的一个重要作用是清除衰老细胞。然而,大多数自发性SLE是由更复杂的免疫因素引起的,而不仅是由一个基因缺陷所决定。SLE发病相关的蛋白质可能的活性作用在SLE中缺陷抗原清除自身抗原和免疫复合物的清除补体相关
24、蛋白:C1q,C1r和C1s,C4C2,血清IgM保护或降解DNA或染色体血清淀粉样P复合物DNA酶1诱导免疫耐受维持淋巴细胞激活的阈值检测自身反应性淋巴细胞Fas和Fas配体,细胞周期抑制剂P21器官特异性自身免疫肾脏疾病FcRB、多态性图13.7影响SLE发病过程中的三类功能蛋白编码基因第一类为清除细胞碎片或保护它们不被免疫识别的蛋白质;第二类为调节淋巴细胞激活或细胞清除的蛋白;第三类为影响器官特异性炎症反应的蛋白。SLE的另一类易感基因是编码调节T或B淋巴细胞耐受或激活阈的基因,例如Fas、Fas配体、信号分子SHP-1、B细胞抑制型受体CD22、FcRIIB和细胞周期抑制蛋白p21。第
25、三类基因编码参与免疫复合物介导的炎症反应的蛋白。例如,具有多态性的FcRIIa和FcRIII,它们的多态性变异体与循环免疫复合物的结合能力不同,因而与SLE中的肾炎发生有关。与SLE易感性有关的另一个重要因素是机体的激素状态。实际上,很多自身免疫病都有明显的性别差异(见图13.3)。在实验动物上也发现自身免疫病的性别差异,如果阉割雄性动物或给予雄性动物雌激素,则能消除性别因素引起的自身免疫病差异。进一步研究发现,自身免疫病的发病高峰正好是女性激素(雌激素和孕激素)分泌旺盛期。深入了解遗传因素和激素效应对自身免疫病的影响,有助于预防自身免疫反应。13-5 抗体和T细胞引起组织损伤导致自身免疫病自
26、身免疫病是由自身抗原引发的针对机体自身的免疫反应,此时,获得性免疫不能清除自身抗原,却形成针对自身抗原的正反馈反应,最后导致组织损伤。IDDM是个例外,因为自身免疫反应将靶器官完全毁损后,将不再有自身抗原胰岛素产生。胰岛素缺乏最后导致糖尿病。自身免疫引起组织损伤的机制可以根据超敏反应来分类(见图13.1和12.2)。与超敏反应相同,效应B细胞和T细胞导致组织损伤。自身免疫病的病理过程及临床表现是由引发自身免疫的自身抗原,以及自身免疫引起组织损伤的机制所决定(见图13.1)。虽然IgE 介导的I型超敏反应似乎不参与自身免疫病,也没有证明IgE抗体介导免疫疾病,但是确实在某些自身免疫病患者体内检测
27、到IgE自身抗体。例如,哮喘、噬酸性红细胞增多症以及罕见的自身免疫性脉管炎(hurg-Strauss vasculitis)都有这种现象。大多情况下,自身免疫与II型超敏反应引起组织损伤的机制相同,即由与细胞膜表面或细胞外基质中的自身抗原结合的IgG或IgM引起组织损伤。自身免疫引起组织损伤的另一种机制与III型超敏反应相同,是由自身抗原与自身抗体形成的循环免疫复合物沉积引起,以自身免疫性血管炎为特征,并且多数是系统性症状。最后,在一些组织特异性自身免疫病中,自身反应性T细胞直接引起组织损伤。对于多数自身免疫病,上述免疫病理机制起协同作用。例如,在SLE、IDDM、类风湿型关节炎时,自身反应性
28、T细胞与抗体都引起组织损伤。下面我们依次讨论自身反应性抗体与自身反应性T细胞引起组织损伤的机制。13-6 针对血细胞的自身抗体使其破坏针对血细胞表面自身抗原的IgG或IgM能够破坏这些细胞。例如,在自身免疫性红细胞溶血症(autoimmune hemolytic anemia)中,针对红细胞表面抗原的自身抗体可使红细胞破裂导致溶血,途径有两条(见图13.8)。在脾脏,单核巨噬系统通过Fc受体和补体受体识别IgG或IgM,清除与自身抗体IgG或IgM结合的红细胞;另一途径是膜攻击复合物溶解自身抗体敏感的红细胞。在自身免疫性血小板减少性紫癜(autoimmune thrombocytopenic
29、purpura)中,针对血小板表面纤维蛋白原GpIIb:IIIa的自身抗体能导致血小板减少而引起出血。红细胞中加入抗红细胞抗体(图)单核巨噬系统中补体受体阳性的细胞吞噬作用和红细胞破坏(图)(图)补体激活和血管内溶血溶细胞作用和红细胞破坏图13.8针对细胞表面抗原的特异性抗体可引起细胞损伤例如,在自身免疫性溶血性贫血中,结合IgG型自身抗体的红细胞被单核巨噬系统表面具有Fc受体的巨噬细胞吞噬(左图),结合IgM型的细胞在锚定C3后被具有CR1或CR3的巨噬细胞清除。这些红细胞的吞噬和清除主要发生在脾脏。红细胞表面结合的某些自身抗体锚定膜攻击复合物的能力比较强,因而引起血管内溶血(右图)。在有核
30、细胞,补体调节蛋白保护这种细胞不容易被补体系统溶解,这些调节蛋白可以干扰补体系统的激活以及妨碍膜攻击复合物的形成(见2-14)。即使补体系统被膜结合抗体激活,有核细胞仍可以通过胞吞或胞吐将形成的膜攻击复合物清除。虽然如此,结合自身抗原的有核细胞仍可能被单核巨噬系统破坏,例如针对嗜中性粒细胞的自身抗体可以引起中性白细胞减少症,从而容易发生化脓性细菌感染。由于脾脏是清除红细胞、血小板以及白细胞的主要器官,因此切除脾脏是治疗自身免疫病的手段之一。13-7 低于溶血剂量的补体锚定于细胞表面可以在组织中引起强烈的炎症反应在组织中,与机体细胞结合的IgG或IgM通过补体引起炎症反应。虽然有核细胞可以抵抗补
31、体的溶细胞作用,但是没有达到溶解细胞水平的膜攻击复合物能引起强烈的炎症反应。在不同的细胞,微量的膜攻击复合物与细胞膜的相互作用引起不同的反应,包括释放细胞因子、前列腺素、白三烯前体花生四烯酸和化学趋化因子等,以及氧爆发(呼吸爆发)和膜脂运动,从而趋化炎性细胞进入到组织。这些趋化分子包括自身抗体激活补体系统释放的补体片段C5a,以及自身抗体激活细胞释放的白三烯B4。趋化到组织的炎性细胞进一步被Fc段和补体C3片段激活,然后与抗体依赖的细胞毒NK细胞一起,进一步加重组织损伤。在Hashimoto氏甲状腺炎(慢性甲状腺炎)(Hashimotos thyroiditis)中,可以持续检测到高滴度针对甲
32、状腺过氧化物酶和甲状腺球蛋白的抗体。下面将讨论直接由T细胞介导的细胞毒作用也是该病发展的重要因素。13-8 针对受体的自身抗体通过刺激或者阻断受体功能而引发疾病当自身抗体和细胞表面的受体结合,可导致一种特殊类型的II型过敏反应,原因这种结合刺激或抑制了该受体的天然配体对它的作用。在Graves氏病(Graves disease)中,甲状腺细胞上的促甲状腺素受体自身抗体可刺激过量的甲状腺素产生。正常情况下,甲状腺素一般是反馈调节的,高水平的甲状腺素抑制垂体促甲状腺素的释放。在Graves氏病中,因为促甲状腺素的缺失,自身抗体持续刺激促甲状腺素受体,使其反馈抑制失调,病人表现为甲亢(图.13.9)
33、。垂体分泌的甲状腺刺激激素(TSH)促进甲状腺释放甲状腺素自身免疫B细胞产生针对TSH受体的抗体,这些抗体可以刺激甲状腺素的产生(图)pituitary:垂体thyroid follicle:甲状腺滤泡thyroid hormoned:甲状腺素(图)甲状腺素可以下调TSH的表达,进一步抑制甲状腺素的合成(负反馈)甲状腺素仍然可以下调TSH的表达,但是对自身抗体的产生没有影响;这些抗体继续刺激过量的甲状腺素产生(图)(图)图13.9在Graves氏病中,甲状腺素产生的反馈调节被破坏Graves氏病是由针对甲状腺刺激激素(TSH)受体的自身抗体引起。正常情况下,甲状腺素的产生是受脑垂体分泌的TSH
34、调控(左图)。在Graves氏病时,针对TSH的自身抗体是其激动剂,因而可以刺激甲状腺素的分泌(右图);甲状腺素虽然可以抑制垂体TSH的产生,却不能影响自身抗体的产生。过量的甲状腺素则引起甲状腺功能亢进。神经肌肉接头处的肌肉细胞表达乙酰胆碱受体。在重症肌无力(myasthenia graves)中,产生针对该受体a链的自身抗体,能阻断神经肌肉的信号传导。自身抗体可导致细胞内和细胞间的乙酰胆碱受体发生降解(图13.10)。重症肌无力患者因为患有自身免疫病,在胎儿时期起就表现出逐渐进展的肌无力症状。自身抗体作为细胞表面受体拮抗剂或促进剂导致的疾病见图13.11。神经肌肉接头处的正常事件重症肌无力(
35、图)神经脉冲(图)神经脉冲乙酰胆碱受体;肌肉;Na+内流引起肌肉收缩乙酰胆碱受体内化并降解;没有Na+内流,肌肉不收缩图13.10在重症肌无力中,自身抗体抑制受体的功能正常情况下,运动神经元释放的乙酰胆碱激动神经肌肉接头处的乙酰胆碱受体,引起肌肉收缩(左图)。在重症肌无力时,针对乙酰胆碱受体亚基的自身抗体不仅不激动受体,反而引起受体的内吞和降解(右图)。由于神经肌肉接头处的受体减少,因此对乙酰胆碱的反应能力下降。针对细胞膜表面受体的自身抗体引起的自身免疫病病症抗原后果Graves氏病甲状腺刺激激素受体甲亢重症肌无力乙酰胆碱受体渐进性肌无力胰岛素抵抗性糖尿病胰岛素受体(拮抗剂)高糖、酮酸中毒低糖
36、血症胰岛素受体(激动剂)低糖血症图13.11针对细胞表面受体引起的自身免疫病自身抗体可作为激动剂或拮抗剂引起不同的效应。注意:针对胰岛素受体的自身抗体既可作为激动剂又可作为拮抗剂。13-9 细胞外抗原的自身抗体通过类似于II型和III型超敏反应的机制导致炎症损伤针对细胞外基质的抗体不常见,一旦出现,损伤将很严重。Goodpasture氏综合征(Goodpastures syndrome)是一种II型过敏反应(见图12.2),生成了针对基底膜胶原(IV型胶原)3链的抗体。该抗体既可与肾小球基底膜结合(图13.12a),又可与肺泡基底膜结合;如果不采取措施,患者将迅速发病。这种自身抗体与基底膜Fc
37、受体结合,活化单核细胞、中性粒细胞、嗜碱性细胞等释放趋化因子,吸引更多的中性粒细胞聚集到肾小球内,导致严重的组织损伤(图13.12b)。这些自身抗体也可激活局部补体,强化组织损伤。图13.12针对肾小球基底膜的自身抗体引起的肾小球炎性疾病称为Goodpasture氏综合症图示Goodpasture氏综合症病人肾小球的连续切片。图a:免疫荧光显示肾小球IgG沉积,针对肾小球基底膜的自身抗体线型沉积在肾小球基底膜,自身抗体引起局部具有Fc受体的细胞和补体的激活,以及中性粒细胞的浸润。图b:伊红、苏木素染色显示肾小球内新生的新月体(C)、肾小囊(bowmans capsule)内增生的单核细胞(B)
38、,肾小球微血管网内中性粒细胞浸润(N)。该图经M.Thompson和D.Evans授权。当抗体和水溶性抗原结合时,则形成免疫复合物(见附录I,A-8部分)。一般来说,具有补体受体的红细胞,和具有补体受体及Fc受体的单核吞噬系统中的吞噬细胞会有效的清除这些复合物,不会引起很大的组织损伤。然而,该清除系统在三种情况下可能会失效。一种情况是给予大量抗原,免疫复合物形成过多,超出了正常的清除能力。血清病就是这样一个例子,它是由于输入了大量的血清蛋白造成的,该疾病持续时间很短,免疫复合物被清除后即可痊愈。另一种情况是慢性感染性疾病,如细菌性心内膜炎,机体针对心房内菌斑的免疫反应不足以消除感染。在强烈的抗
39、细菌抗体作用下,心房内持续的细菌抗原释放会导致肾脏、皮肤等器官小血管广泛发生免疫复合物损伤。第三种见于SLE,它是一种免疫复合物介导的疾病,主要是产生了针对有核细胞多种自身抗原的IgG抗体。这些抗原为细胞内的三种核蛋白核小体、结合体和胞浆核蛋白复合物,包括Ro和La蛋白(以首次发现该蛋白自身抗体的患者姓氏的前两字母命名)。这些自身抗原转移到细胞外形成免疫复合物。在SLE中,死亡和接近死亡的细胞,以及受损的组织释放自身抗原,大量的微小免疫复合物持续产生,沉积在肾小球、肾小球基底膜(图13.13)和关节等器官的小血管壁上,随后导致吞噬细胞活化。后续的组织损伤释放更多的核蛋白复合物,产生更多的免疫复
40、合物。最后,小血管壁上的炎症反应,尤其是肾脏的病变,将导致病人死亡。缺乏Fc RIII的小鼠尽管也有免疫复合物和C3的沉积,但是不会产生肾小球肾炎,表明在SLE自身免疫机制中,Fc受体起了很重要的作用。图图13.13在SLE,由于免疫复合物在肾小球沉积引起肾功能衰竭图a:SLE患者的肾小球切片,可以看到免疫复合物沉积引起基底膜增厚(肾小球内出现的裂隙);图b:免疫荧光显示免疫球蛋白沉积在肾小球基底膜;图c:电镜显示在肾小球基底膜和肾小球上皮细胞间高密度的免疫复合物沉积,以及趋化而来的多形核中性粒细胞。该图由M.Kashgarian授权。13-10环境协同因子影响自身免疫病的表现只有自身抗体和自
41、身抗原结合后,自身抗体才引起自身免疫病。以下两个例子说明自身抗原和环境辅助因子影响疾病的表现。在未治疗的Goodpasture氏病中,IV型胶原的自身抗体可引起致命性肾小球肾炎。IV型胶原广泛分布于全身的基底膜,包括肺泡、肾小球及内耳耳蜗。所有的Goodpasture氏病患者均有肾小球肾炎,40%有肺出血,但不发生耳聋;几乎所有吸烟的病人都会出现肺出血。这些现象是由肾小球、肺泡和耳蜗基底膜的不同导致抗原抗体结合的效率出现差异。肾小球基底膜的功能主要是滤过血浆。肾小球毛细血管的内皮细胞是有孔的,血浆可以通过这些空隙与基底膜接触,也就意味着循环中的自身抗体很容易接触肾小球基底膜。在肺泡则相反,肺泡
42、毛细血管的内皮细胞紧密连接,基底膜将肺泡内皮与毛细管内皮分开。如果抗体要到达基底膜,则必须有肺泡毛细血管内皮连接发生损伤。吸烟可以刺激肺部出现炎症反应,损害肺泡毛细血管,使得自身抗原与抗体接触。而耳蜗基底膜似乎在任何时候都不会接触到自身抗体。另外一个说明环境影响自身免疫病症状的例子是感染对Wegener氏肉芽肿性脉管炎(Wegeners granulomatosis)的影响。该病表现为严重的坏死性脉管炎(图13.14),由一种抗中性粒细胞浆抗体(anti-neutrophil cytoplasm antibodies,ANCA),即中性粒细胞颗粒蛋白酶自身抗体引发。其对应的自身抗原是蛋白酶-3
43、(proteinase-3),为一种中性粒细胞丝氨酸蛋白酶。一般认为,ANCA的水平与疾病的表现有关,但是也经常见到高水平ANCA患者无任何症状。如果患者出现感染,则常常出现严重的脉管炎。原因是静止的中性粒细胞表面不表达蛋白酶-3,无感染时抗原不会接触到抗蛋白酶-3自身抗体。感染后,多种细胞因子激活中性粒细胞,蛋白酶-3转位至细胞表面,抗蛋白酶-3抗体与之结合后刺激中性粒细胞脱颗粒和释放自由基。同时,感染活化的血管内皮细胞可表达血管粘附分子(如E-选择素),促进活化的中性粒细胞结合到血管壁上,对其造成损伤。通过这种途径,很多非特异的感染使自身免疫病出现恶化。图图13.14在Wegener氏肉芽
44、肿病人的血清中,存在与中性粒细胞胞浆颗粒反应的自身抗体增加了细胞膜通透性的中性粒细胞与Wegener氏肉芽肿病人的血清孵育后,用标记荧光的抗IgG 的抗体检测血清中与中性粒细胞胞浆颗粒反应的抗体。该图由C.Pusey授权。13-11自身免疫中的炎症损伤方式可以因为解剖学的限制而得以改善在Goodpasture氏综合症中,损伤发生于什么器官取决于基底膜胶原能否有效接触到自身抗体;同时,环境因素也可影响不同器官中抗原接触抗体的有效性。膜性肾小球肾炎(membranous glomerulonephritis)可以解释解剖因素对免疫性炎症的影响,该病患者有严重的蛋白尿(尿中出现多余的蛋白),当血浆蛋
45、白下降到一定的程度后,即可危及生命。肾活检发现患者的肾小球基底膜有抗体和补体沉积。与Goodpasture氏综合症相反的是,该病没有明显的炎症细胞浸润,致病的自身抗原也未确定。但是,通过注射肾小管上皮组织,可以诱导产生针对肾小管上皮细胞糖蛋白的自身抗体,从而制备出很好的啮齿类膜性肾小球肾炎模型Heymann氏肾炎(Heymanns nephritis)。膜攻击补体复合物中的蛋白损耗可以使蛋白尿消失,但是中性粒细胞的损耗不会出现同样的效果。该现象表明,沉积在肾小球基底膜下的抗体通过激活补体而引起组织损伤,而肾小球基底膜充当了炎症反应白细胞的补体载体。在其它自身免疫病中,即使没有抗体介导的炎症反应
46、,仍然可有高水平的抗细胞内抗原的自身抗体。一种少见的伴有肺纤维化的肌炎就是这样一个例子。很多患有该病的患者具有高水平的氨酰-tRNA合成酶自身抗体,这种酶能将tRNA连接到氨基酸上。在体外培养的细胞中加入这些自身抗体,可以完全阻断翻译和蛋白合成。然而,没有任何证据表明这些抗体能在体内造成伤害,因为它们无法进入细胞内。该病的自身抗体一般作为一种特殊类型的组织损伤标记物,并不参与肌炎的免疫病理。其它类似的自身抗体还有原发性胆汁硬化症中的抗线粒体抗原抗体,慢性活动性肝炎时的抗平滑肌抗原抗体。13-12自身免疫引起组织损伤的机制可通过过继性转移来确定在确定某种疾病是自身免疫病之前,我们必须确定它的病理
47、过程是由针对自身抗原的自身抗体引起。起初,大家认为只要在患者血清中测到自身抗体,就认为该病的发生是以自身免疫为基础。但是,在外伤和感染时同样可以检测到低亲和力的抗体,这表明自身抗体既可以是组织损伤的原因,也可以是组织损伤的结果。因此,在确定某个自身抗体引发自身免疫病前,必须明确该抗体的病理过程。我们可以将患者血清中的抗体注射到动物体内,获得病理过程相同的动物模型(见图13.15)。然而,由于种属间抗原结构的差异,这种造模方法有时会失败。有些自身免疫病可以从患病的母体传给胎儿,也就是说,新生儿可以患与母亲同样的自身免疫病(见图13.16)。这种现象是由于自身抗体IgG穿过胎盘引起的(见图13.1
48、7),患儿的症状随着母体抗体的代谢很快消失,该现象很好地解释自身抗体是致病因素。但母亲患SLE或Sjgren氏综合征时,却能引起新生儿慢性心脏损伤。在没有发生先天性心脏阻滞(congenital heart block)前,可以通过全血或血浆置换进行治疗。Graves病患者的血清中含有针对甲状腺刺激激素受体的抗体(图)将Graves病患者的血清注入大鼠体内(图)甲状腺素的水平升高图13.15自身免疫病人的血清可将这种疾病转移到实验动物当人、小鼠或大鼠的自身抗原非常相似时,自身免疫病人的自身抗体可以在实验动物身上引起相同的症状。例如,Graves氏病人的自身抗体经常可在实验动物身上引起相同的症状
49、。透过胎盘引起胎儿或新生儿的自身免疫病疾病抗体症状重症肌无力抗乙酰胆碱受体抗体肌无力Graves病抗甲状腺刺激激素受体抗体甲亢血小板减少性紫癜抗血小板抗体淤斑、出血新生儿红斑狼疮和先天性心脏传导阻滞抗Ro抗体抗La抗体光敏性淤斑和/或心动过缓寻常性天疱疮抗桥粒芯糖蛋白-3抗体水疱性皮疹图13.16透过胎盘的自身反应型IgG抗体可以引起胎儿或新生儿的自身免疫病这些疾病主要是由针对细胞表面和组织基质中的分子。这种现象提示,决定透过胎盘的IgG抗体是否可以引起胎儿或新生儿的自身免疫病的重要因素是自身抗原是否可以和自身抗体接触。先天性自身免疫性心脏传导阻滞是由心脏传导系统的纤维化引起的心动过缓。实验表
50、明这种病人表达过量的Ro抗原(见13-9)。Ro蛋白是细胞内小分子的核糖体蛋白。现在还没有证据表明这种蛋白可以在细胞表面或心脏传导系统中表达,从而作为自身免疫病引起组织损伤的靶位点。Graves病患者产生针对甲状腺刺激激素受体的抗体这些抗体可以通过胎盘进入胎儿体内新生儿同样罹患Graves病血浆置换法可以除去母体来源的针对甲状腺刺激激素受体的抗体,治疗该病(图)(图)(图)(图)图13.17由于抗体可以透过胎盘,新生儿出现母体罹患的抗体介导的自身免疫病如图9.22所示,IgG型抗体可以透过胎盘在胎儿体内聚集。因而在出生后几周内,新生儿可以出现与母体罹患的自身免疫病相同的症状。幸运的是,随着母体
51、自身抗体的清除,这些症状可以很快消失,不会引起持续的组织损伤。在Graves氏病,疾病症状是由针对甲状腺刺激激素(TSHR)的抗体引起,产生TSHR抗体的母亲娩出的胎儿出现甲亢,这些症状可以通过血浆置换清除母体抗体而改善。13-13针对自身抗原的特异性T细胞不仅直接引起组织损伤,而且参与持续的自身抗体应答针对自身抗原肽/MHC复合体的效应T细胞可以直接或激活单核巨噬细胞后间接引起组织局部的炎症反应。这种T细胞参与IDDM、类风湿性关节炎、多发性硬化的发生发展,受累组织可以见到大量的T淋巴细胞和单核巨噬细胞浸润。针对自身MHC分子呈递的自身抗原特异性T细胞不仅介导这些自身免疫病,而且维持所有的自
52、身抗体反应。证实自身反应性T细胞要比证明自身抗体参与免疫反应困难得多:(1)我们不能把自身反应性T细胞转移到动物造成疾病模型,因为T细胞识别抗原受MHC限制,而人类和动物的MHC等位基因不同;(2)很难确定自身反应T细胞识别的自身抗原。例如,我们可以通过免疫组化确定产生自身抗体的抗原组织分布,而自身反应T细胞则无法确定。但是,也有证据证明自身反应性T细胞确实参与了自身免疫病。例如IDDM时,产生胰岛素的胰岛b细胞是由自身反应性T细胞选择性毁损。当将患者同卵双生子的胰腺移植过来,CD8 T细胞很快有选择性地把供者的b细胞破坏。给予T细胞激活抑制剂环孢霉素A可以预防该病的复发。在13-15中,我们
53、将进一步讨论自身反应性T的鉴定标记,以及这些细胞引起自身免疫病的证据。13-14自身抗体可用来确定自身免疫进程的靶抗原可以用自身抗体来纯化和鉴定自身抗原,尤其是当自身抗体能够造成动物模型时,有助于获得大量的组织标本。另外,免疫组化能够确定产生自身抗原的细胞与组织分布,为疾病的病理过程分析提供线索。在鉴定自身抗原的过程中,同样有助于确定刺激抗体产生的CD4 T细胞。第八章已提到,CD4 T细胞选择性激活B细胞,后者可以与T细胞识别的肽抗原决定簇结合。因此,用自身抗体纯化的蛋白或蛋白复合体包含有自身反应性CD4 T细胞结合肽。例如,重症肌无力中乙酰胆碱受体的自身抗体可以用来从肌肉组织的匀浆中纯化乙
54、酰胆碱受体,并可用于发现与该受体亚基肽段结合的CD4 T细胞(见图13.18)。因此,自身反应性B细胞和T细胞都参与该病的病理过程。从重症肌无力病人分离全血分离的血清中含有自身抗体将肌肉组织匀浆与分离的血清免疫共沉淀证明自身抗体的靶抗原是乙酰胆碱受体图图图外周血单核细胞中含有T细胞从病人血清中可以培养出特异性针对乙酰胆碱受体的T细胞图图图13.18自身抗体引起的自身免疫病需要自身反应性T细胞从graves氏肌无力病人血清中分离的自身抗体可以与骨骼肌匀浆中的乙酰胆碱受体发生免疫共沉定(上图)。为能产生自身反应性抗体,必须存在与乙酰胆碱受体抗原肽反应的CD4阳性T细胞。为检测这些细胞,可从grav
55、es氏肌无力病人血清中分离T细胞,与相同MHC组织型的抗原递呈细胞和乙酰胆碱受体共培养(下图)。只有与乙酰胆碱受体特异表位反应的T细胞才能被刺激增殖。SLE也同样存在这种现象,其多种核蛋白复合体的自身抗体都可引起组织损伤(见13-9),并且显示出高度的体细胞突变(抗原刺激抗体产生的一个特征,见4-9),产生这些抗体的B细胞发生高度的克隆扩增,由此进一步证明,自身抗体是由特异的自身抗原刺激,并在特异的CD4阳性T细胞辅助下产生。SLE患者的自身抗体可以结合核蛋白复合物的任一组分,显示抗原是CD4阳性T细胞能够识别的肽段。如图13.19,所有识别自身核蛋白复合物的B细胞把自身抗原内吞和加工后,将自
56、身抗原肽呈递给自身反应性CD4阳性T细胞,并接受来自辅助T细胞的信息。这种B细胞与T细胞的相互作用,引发了B细胞分泌自身抗体、克隆扩增和体细胞高频突变,是产生自身抗体反应特征性表现和患者自身抗体特异性的聚集效应的原因,使得针对多分子复合物成分产生的自身抗体发生扩散,也称作抗原扩展(antigen spreading)或抗原决定簇扩展(determinant spreading)。组蛋白H1特异性辅助T细胞激活组蛋白H1特异性B细胞,这种B细胞加工含有H1组蛋白的核小体并呈递H1的抗原肽激活的B细胞分化为浆细胞产生针对组蛋白H1的抗体(图)核小体H1特异性T细胞B细胞(图)组蛋白H1特异性辅助T
57、细胞激活DNA特异性B细胞,这种B细胞加工核小体并呈递H1的抗原肽激活的B细胞分化为浆细胞产生针对DNA的抗体(图)(图)组蛋白H1特异的辅助T细胞不能激活核糖体特异的B细胞,这些B细胞识别核糖体并呈递核糖体特异的抗原肽在没有核糖体特异的T辅助细胞存在的情况下,这些B细胞不能激活,没有抗体产生(图)核小体(图)图13.19由于抗原扩展某一特异性的辅助T细胞,可以促使产生几种特异性的自身抗体例如,在SLE病人,特异识别核小体中组蛋白H1的B细胞可以将核小体内吞,将来自H1和核小体其它组分的抗原肽呈递到其表面,这种B细胞收到来自特异识别H1抗原肽的T细胞辅助可以活化增殖(最上图)。特异识别核小体上D
温馨提示
- 1. 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
- 2. 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
- 3. 本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
- 4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
- 5. 人人文库网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
- 6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
- 7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
最新文档
- 2025年高职国际航运业务管理(航运业务操作)试题及答案
- 2025年高职航空机电设备维修(航空设备维护)试题及答案
- 2025年高职(食品生物技术)食品酶制剂应用专项测试试题及答案
- 2025年高职生态保护运营(运营技术)试题及答案
- 2025年大学戏剧影视表演(表演基础)试题及答案
- 2025年高职(智能制造装备技术)装备维护阶段测试题及答案
- 2025年高职(给排水工程技术专业)管道维修试题及答案
- 2025年大学休闲体育(康乐体育)试题及答案
- 2025年高职地理教育(地理教学设计)试题及答案
- 2025年高职(园林技术)绿化工程施工实训试题及答案
- 苏州市姑苏区教育体育和文化旅游委员会下属学校招聘事业编制教师笔试真题2023
- 后切式背栓连接干挂石材幕墙施工方案
- 人教版数学四年级上册期末测试卷及答案 (共八套)-2
- 大转炉氧枪橡胶软管和金属软管性能比较
- 四川省内江市2023-2024学年高二上学期期末检测生物试题
- 02-废气收集系统-风管设计课件
- 2022ABBUMC100.3智能电机控制器
- 天津东疆我工作图0718
- GB/T 19367-2022人造板的尺寸测定
- 北京春季化学会考试卷及答案
- 数学建模插值与拟合
评论
0/150
提交评论