

下载本文档
版权说明:本文档由用户提供并上传,收益归属内容提供方,若内容存在侵权,请进行举报或认领
文档简介
1、分子内坐标输入以含 H2O 的高斯输入格式为例:# p HF/STO-3GWater energy 0 1OH 1 0.956H 1 0.956 2 104.5 计算的最基本格式如上例所示,第一行表示计算方法和基组,然后空一行, 第三行表示名称, 然后空一行, 蓝色的一行两个数字分别表示分子电荷和自旋 多重度,红色部分为分子内坐标。在分子内坐标系中, 分子中每个原子的相对位置是用与它成键的另一原子间键长、 该键 与另一化学键间的键角, 以及后者与和它有一条公共边的另一键角所成的二面角来确定。 可 以理解为原子在空间的相对位置, 取分子中的一个原子作为参考原子逐步确定其他原子的位 置。需注意:键
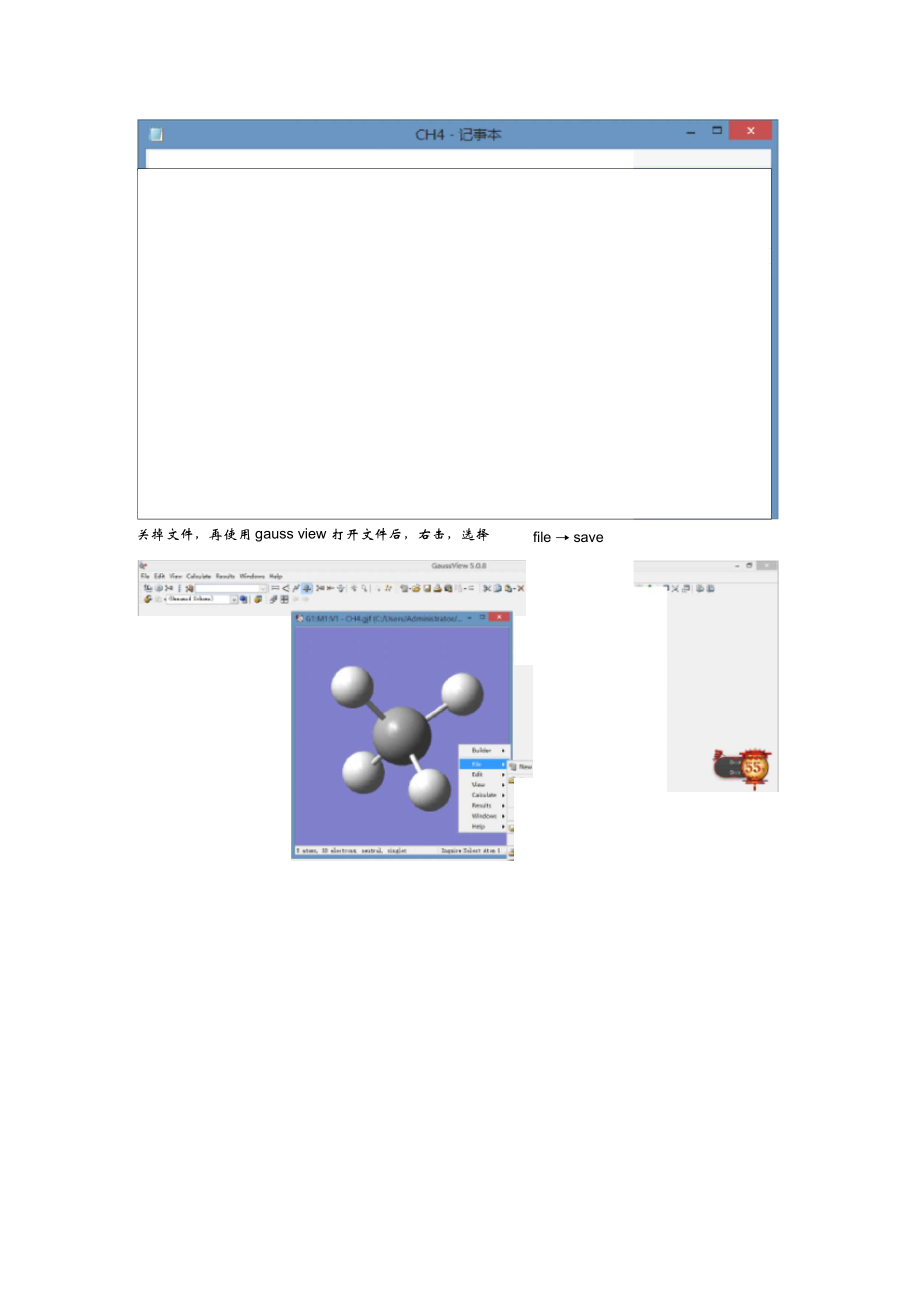
2、长 的默认单位为 ?;0o< 键角 180o。二面角 的定义为包含原子 A、1、2 的平面与包含原子 1、2、 3 的平面所成的夹角, 其取值区间为 -180o, 180o。二面角所取的正负符号由 “右旋法则 ”确定。用“右旋法则” 确定二面角的正负时,取包含参考 3 个原子的平面为基准面、 参考原子2 指向参考原子 1 的位矢方向为基转轴的正方向。若被定义的原子 A 与参考原子 1、 2 构成 的平面位于基准面的逆时针方位,则其二面角参量为正号,否则为负号。以上是关于内坐标的解释,我们在输入分子内坐标时大多依靠软件( chem3D 和 gauss view)进行。下面以甲烷为例说明 :首先使用 chem3D 画一个甲烷分子;然后另存为格式为 gjf使用记事本打开保存的文件,如果是内坐标表示完成;如果显示的是笛卡尔坐标如下file save关掉文件,再使用 gauss view 打开文件后,右击,选择文件储存类型选择 Gaussian Input Files( *gjf ),write Cart
温馨提示
- 1. 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
- 2. 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
- 3. 本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
- 4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
- 5. 人人文库网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
- 6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
- 7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
最新文档
- 人工智能视觉幻象技术
- 幼儿园开学典礼主持词15篇
- 人工基础智能及概论 9
- 2025-2026学年教学设计类的硕士论文
- 2025-2026学年柳荷叶圆圆教案
- 黑龙江省龙东十校联盟2025-2026学年高一上学期期末考试物理试题
- 蓝色学生竞选介绍
- 2025-2026学年《故宫》教学设计看法
- 2025-2026学年教案小池
- 2025-2026学年教学游戏化环节设计
- 出纳员职业技能鉴定考试复习题库(附答案)
- 加油站风险辨识与安全管控培训
- 2025年四川省自贡市地理生物会考真题试卷+答案
- GB 26396-2026洗涤用品安全技术规范
- 2026年上海市宝山区中考一模化学试卷
- 东南大学2024综评数学试卷
- DB31∕T 1545-2025 卫生健康数据分类分级要求
- 广东省安装工程综合定额(2018)Excel版
- 生命哲学:爱、美与死亡智慧树知到期末考试答案章节答案2024年四川大学
- 浙江省衢州市各县区乡镇行政村村庄村名居民村民委员会明细及行政区划代码
- 防洪防汛安全培训记录
评论
0/150
提交评论